Вода — весьма своеобразный
растворитель. Прежде всего — она кипит и затвердевает при аномально
высоких, для ее малого молекулярного веса, температурах. Действительно,
вода — Н2О — кипит
при 373оК, а твердеет при 273оК,
в то время как О2 кипит при 90оК
и твердеет при 54оК; Н2
кипит при всего 20оК и плавится при 4
оК; СН4 кипит при 114оК,
и т.д. То, что структуры воды и льда с трудом разрушаются теплом, показывает,
что молекулы воды чем-то очень сильно связаны между собой.
Это "что-то" — связь
между О и Н атомами молекул Н2О (именно
между О и Н, так как и О2, и Н2
в отдельности легко кипят и плавятся). Эта связь называется водородной
связью.
Водородные связи наблюдаются не
только в воде. Они наблюдаются всегда, когда водород химически связан с
одним электроотрицательным (т.е. притягивающим электрон) атомом и при этом
приближается к другому электроотрицательному атому. Примеры: O—H : : :
O, N—H : : : O, N—H : : : N. Но С-H группа, например, водородных связей
не образует: атом С — недостаточно электроотрицательный атом.
Основные особенности воды как
растворителя связаны с наличием в ней мощных водородных связей.
Водородная связь молекул воды носит
электрическую природу. То, что она связана именно с электронами и зарядами,
а не с ядрами водородных атомов, прямо следует из того, что температуры
и теплоты кипения и плавления легкой (Н2О)
и тяжелой (D2О) воды практически совпадают,
— несмотря на двукратное различие в массах ядер D и Н.
Молекула воды полярна. Это значит,
что на ее атомах есть небольшие ("парциальные") электрические заряды: на
электроотрицательном О — отрицательный, на Н —
положительный. Распределение зарядов и электронных облаков на этих атомах
выглядит так:
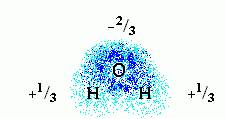
Здесь плотность электронного облака
показана плотностью точек, а цифры показывают парциальный заряд атомов
этой полярной молекулы. Заряд этот выражен в долях от протонного заряда.
У протона, в этих единицах, заряд, естественно, равен +1, а у электрона
-1. В целом молекула воды имеет заряд
0.
Заряды на полярных атомах появляются
в результате того, что электроотрицательные атомы O оттягивают электронные
облака от соседних Н атомов. В результате на последних возникают небольшие
положительные заряды, а на O — отрицательный заряд.
Как вы помните, заряды q1
и q2 на расстоянии r взаимодействуют
в вакууме с энергией
| U = q1q2/r. | (4.1) |
В вакууме заряды взаимодействуют
очень сильно. Энергия взаимодействия двух единичных (т.е. протонных или
электронных) зарядов на расстоянии 1![]() близка к 330 ккал/моль (запомните эту цифру: она нам пригодится для разных
дальнейших оценок), а на более реалистичном (учитывая Вандерваальсово отталкивание
атомов) расстоянии 3
близка к 330 ккал/моль (запомните эту цифру: она нам пригодится для разных
дальнейших оценок), а на более реалистичном (учитывая Вандерваальсово отталкивание
атомов) расстоянии 3![]() эта энергия близка к 110 ккал/моль. Энергия взаимодействия единичных зарядов
— это характерная энергия химической связи; она в сотни раз больше,
чем характерная тепловая энергия kT или характерная энергия Вандерваальсового
взаимодействия атомов.
эта энергия близка к 110 ккал/моль. Энергия взаимодействия единичных зарядов
— это характерная энергия химической связи; она в сотни раз больше,
чем характерная тепловая энергия kT или характерная энергия Вандерваальсового
взаимодействия атомов.
Парциальные заряды молекулы воды
меньше и потому взаимодействуют слабее — с энергией порядка
10 ккал/моль на расстоянии 3![]() ,
но и такой энергии хватает на то, чтобы, при притяжении Н к О, "промять"
электронные оболочки атомов. Особенно этот эффект "проминания" облаков
существенен для атомов водорода: здесь электронная оболочка Н, и так состоявшая
всего из одного электрона, оттянута к О атому, так что смять ее особенно
легко. Смять электронную оболочку, скажем, О атома много труднее
— вокруг ядра О вращается 8 "своих" электронов, да еще какую-то часть
электронного облака атом О (в Н2О) оттягивает
у соседних водородов.
,
но и такой энергии хватает на то, чтобы, при притяжении Н к О, "промять"
электронные оболочки атомов. Особенно этот эффект "проминания" облаков
существенен для атомов водорода: здесь электронная оболочка Н, и так состоявшая
всего из одного электрона, оттянута к О атому, так что смять ее особенно
легко. Смять электронную оболочку, скажем, О атома много труднее
— вокруг ядра О вращается 8 "своих" электронов, да еще какую-то часть
электронного облака атом О (в Н2О) оттягивает
у соседних водородов.
Легкость проминания электронного
облака у Н атома и превращает обычное электростатическое взаимодействие
в водородную связь. Это относится ко всем водородным связям, из которых
нас будут интересовать следующие, встречающиеся в белках связи: O—H : :
: O, N—H : : : O, N—H : : : N.
Итак, электронное облако при Н
атоме — самое слабое, и оно сильно сминается при притяжении
парциального положительного заряда Н к парциальному отрицательному заряду
O (или N) атома. В результате ядро Н подходит к ядру О или N не на 2.35
— 2.75![]() (как то было бы при Вандерваальсовом взаимодействии, о котором мы говорили
на прошлой лекции), а заметно ближе — на расстояние 1.8
— 2.1
(как то было бы при Вандерваальсовом взаимодействии, о котором мы говорили
на прошлой лекции), а заметно ближе — на расстояние 1.8
— 2.1 ![]() (эти данные получены из кристаллов малых молекул).
(эти данные получены из кристаллов малых молекул).
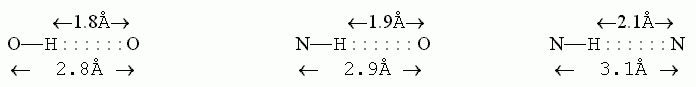
При таком сближении и возникает
водородная связь (или Н-связь). Н-атом (или, точнее, О-Н
или N-Н группа) называется донором водородной связи, а тот О или
N атом, к которому этот Н приближается, называется акцептором водородной
связи.
Обратите внимание, что наблюдающееся
в кристаллах при образовании Н-связи расстояние между О и/или N атомами
донора и акцептора близко к 3.0![]() (например, во льду расстояние между ними равно 2.8
(например, во льду расстояние между ними равно 2.8![]() ).
Это соответствует оптимальному Вандерваальсовому расстоянию между О и/или
N атомами — т.е. наличие стоящего между ними Н атома не приводит
к увеличению расстояния между донорным О (или N) и акцепторным О (или N)
атомами: Н атом их не расталкивает, а связывает.
).
Это соответствует оптимальному Вандерваальсовому расстоянию между О и/или
N атомами — т.е. наличие стоящего между ними Н атома не приводит
к увеличению расстояния между донорным О (или N) и акцепторным О (или N)
атомами: Н атом их не расталкивает, а связывает.
У каждой Н-связи один донор и один
акцептор. При этом Н почти всегда выступает донором только одной Н-связи,
а О может быть акцептором двух Н-связей. В последнем случае возникает "вилочковая"
водородная связь.
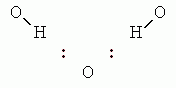
Так как при образовании "вилочки"
сильно сближаются два Н-атома (с зарядом +1/3
каждый), то ее суммарная энергия меньше, чем удвоенная энергия одинарной
(как на предыдущей схеме) Н-связи.
В отличие от Вандерваальсова взаимодействия,
водородная связь весьма чувствительна к направлению — особенно
к направлению донорной группы. Обычно валентная связь донора (N-H или О-Н)
прямо смотрит на тот акцептор (О или N атом), с коим водородная связь образуется:
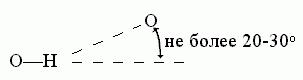
К ориентации акцепторной группировки
водородная связь значительно менее чувствительна:
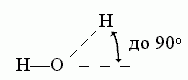
Энергия водородной связи составляет
около 5 ккал/моль. Эта оценка базируется на сравнении экспериментальных
теплот испарения сходных соединений, из которых одни могут завязывать водородные
связи, а другие — нет. Например, теплота испарения диметилэфира
— Н3С-О-СН3
— составляет около 5 ккал/моль, а теплота испарения спирта
— СН3-СН2-ОН
— около 10 ккал/моль. Эти вещества состоят из одних и тех же атомов
— т.е. Вандерваальсовы взаимодействия в них практически одинаковы
— но спирт может завязывать водородную связь, а диметилэфир
— нет (у него нет О-Н группы!). Каждая ОН группа может быть донором
только одной Н-связи, а ее О может такую связь акцептировать. Так как у
каждой Н-связи должен быть один донор и акцептор, в спирте приходится по
одной Н-связи на молекулу, т.е. цена одной водородной связи —
около 10 ккал/моль - 5 ккал/моль = 5 ккал/моль.
Такая же оценка получается из
теплоты испарения льда (680 кал/г = 680 кал/(1/18 моль) = 12 ккал/моль).
Из них пара ккал/моль приходится на Вандерваальсовы взаимодействия
— как видно из испарения малых молекул типа метана (СН4)
или О2. На Н-связи остается около 10 ккал/моль.
Во льду приходится по две водородных связи на молекулу Н2О,
т.к. Н2О, со своими двумя ОН группами,
может быть донором двух Н-связей (и столько же она может акцептировать
своим О атомом). Значит, опять получается — примерно
— (10 ккал/моль)/2 = 5 ккал/моль.
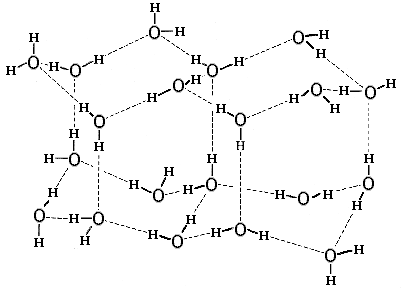
Рис.4-1. Нормальный лед. Пунктир
— Н-связи. В ажурной структуре льда видны небольшие полости, окруженные
молекулами Н2О. Картинка взята из [6].
Структура нормального льда диктуется
Н-связями (Рис.4-1): она хороша для геометрии этих связей (О-Н смотрит
прямо на О), но не очень хороша для плотного Вандерваальсового контакта
молекул Н2О. Поэтому структура льда ажурна,
в нем молекулы Н2О обволакивают микроскопические
(размером меньше молекулы Н2О) поры. Ажурность
структуры льда приводит к двум хорошо известным эффектам: (1) лед менее
плотен, чем вода, он плавает в ней; и (2) под сильным давлением —
например, лезвия конька — лед подплавляется.
Большинство из существующих во
льду (Рис.4-1) водородных связей сохраняется и в жидкой воде. Это следует
из малости теплоты плавления льда (80 кал/г) по сравнению с теплотой кипения
воды (600 кал/г при 0оС). Можно было бы
сказать, что в жидкой воде рвется только 80/(600+80) = 12% из существующих
во льду Н-связей. Однако эта картина — что часть водородных
связей в воде разорвана, а часть сохранена — неточна: скорее,
все водородные связи в воде разбалтываются. Это хорошо иллюстрируется следующими
экспериментальными данными.
На приведенном внизу Рисунке 4-2,
кривая 1 соответствует максимуму инфракрасного спектра поглощения групп
О-Н во льду (где все Н-связи завязаны); кривая 2 соответствует максимуму
инфракрасного спектра поглощения групп О-Н отдельных молекул Н2О,
растворенных в CCl4 (где Н-связей нет
— раствор Н2О в CCl4
слишком разбавлен); а кривая 3 соответствует спектру поглощения жидкой
воды. Если бы в жидкой воде было бы два сорта О-Н групп — образующие
водородные связи и не образующие их — и одни О-Н группы в воде
колебались бы так же (с той же частотой), как во льду (где они образуют
Н-связи), а другие — как в окружении CCl4
(где они Н-связей не образуют). Тогда спектр воды имел бы два максимума,
соответствующие двум состояниям О-Н групп, двум их характерным частотам
колебаний: с какой частотой группа колеблется, с такой и поглощает свет.
Но "двухмаксимумная" картина не наблюдается! Вместо нее на кривой
3 мы видим один, очень размытый максимум, простирающийся от максимума кривой
1 до максимума кривой 2. Это значит, что все О-Н группы в жидкой воде завязывают
водородные связи — но все эти связи разболтаны, причем по-разному.
Это показывает, что картина, в
которой часть водородных связей в воде разорвана, а часть сохранена, -
строго говоря, неверна. Однако она столь проста и удобна для описания термодинамических
свойств воды, что ею широко пользуются — и мы тоже будем к
ней обращаться. Но надо иметь в виду, что она не точна.
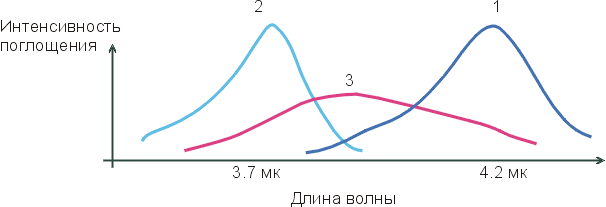
Рис.4-2. Характерный вид инфракрасных
спектров поглощения О-Н групп во льду (1), в окружении CCl4
(2) и в жидкой воде (3).
Обратимся теперь к взаимодействию
белковой цепи с водой.
Как и вода, остов белковой цепи
полярен. Точнее — полярны входящие в остов пептидные группы.
В целом остов белковой цепи имеет заряд 0, но распределение зарядов на
его атомах (здесь опять заряд каждого атома выражен в долях от протонного
заряда) выглядит так:
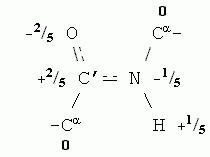
Парциальные заряды содержат и некоторые боковые группы, например, Ser (его боковая группа — -СН2-ОН — похожа на спирт). Еще более полярны заряженные аминокислотные остатки: заряд +1 присущ основаниям, входящим в боковые группы остатков Arg, Lys и His, когда они находятся в ионизованной форме (при примерно нейтральных рН), а у ионизованных кислотных боковых групп, Asp и Glu, заряд равен -1.
И пептидные группы главной цепи, и полярные боковые группы выступают донорами и акцепторами водородных связей. Они могут завязывать водородные связи друг с другом или с молекулами воды — и практически все они завязывают такие связи, так как энергия водородных связей — 5 ккал/моль — раз в десять превосходит энергию теплового движения, т.е. тепловое движение не может разрушить эти связи — разве что время от времени оно случайно нарушает малую часть из них.
Если внутримолекулярная связь между
донором (D) и акцептором (А) водородной связи в белке завязывается в водном
окружении, то эта связь замещает две водородные связи белка с молекулами
воды (которые были до того, т.к. свято место пусто не бывает); и при этом
еще создается одна связь между оторвавшимися молекулами воды:
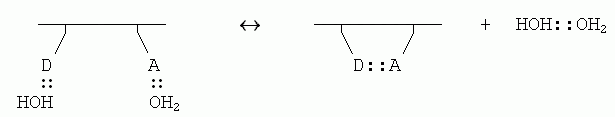
Энергетический баланс этой
реакции близок к нулю: две Н-связи было, две стало. Однако энтропия
воды возрастает, так как вода больше не привязана к белковой цепи и связанные
Н-связью молекулы могут плавать где хочется (а энтропия именно и связана
с множественностью состояний системы). При этом рост энтропии за счет освобождения
димера воды от белка — примерно такой же, как рост энтропии
при уходе молекулы Н2О изо льда в жидкую
воду (в обоих случаях одна частица обретает свободу плавать).
Энтропийный эффект выхода молекулы
Н2О изо льда в воду можно оценить следующим
образом. Как известно, в точке плавления (для льда — при 0оС,
т.е. при 273оК) энтропийный выигрыш от
плавления как раз компенсирует энергетический проигрыш от него. А этот
проигрыш нам известен: 80 кал/г, т.е. 80 Х 18 = 1440 кал/моль (18 - молекулярный
вес Н2О).
Значит, вследствие роста энтропии
свободная энергия системы "белок в воде" падает, — примерно
на 1.5 килокалории на каждый моль формирующихся внутрибелковых водородных
связей D:::A.
Эта выделенная водой свободная
энергия может полностью или частично скомпенсировать рост свободной энергии,
сопровождающий падение энтропии при фиксации групп D и А связью D:::A (раньше
они болтались как хотели, а теперь, связанные, — не могут).
Позже мы увидим, что падение свободной энергии воды примерно компенсирует
энтропию необходимой для образования водородной связи (N-H:::O) фиксации
мономера во вторичных структурах полипептидных цепей. В результате регулярная
вторичная структура полипептидов в воде находится как раз на грани стабильности.
Итак, страшные слова — "энтропия" и "свободная энергия" — произнесены. Мой опыт показывает, что "энтропию" нормальный студент-биолог знает на слух (он слышал, что это — мера множественности состояний, мера беспорядка), а свободную энергию представляет совсем смутно... Так как нам понятие "свободная энергия" понадобится часто, я хочу уделить ей несколько минут сейчас, а потом, когда будет необходимо, поговорить о ней — и об энтропии — еще подробнее.
Начнем с простого примера: пусть
молекула может находиться в двух состояниях, "а" и "б", например,
— здесь, в этой комнате, на высоте 200 м от уровня моря ("а"), и
в монастыре Далай-Ламы, в Гималаях, на высоте 5 км ("б"). Как соотносятся
вероятности этих двух состояний — при условии, что мы следим
за нашей молекулой так долго, что она успеет побывать и здесь, и там?
Обычно все помнят формулу Больцмана,
которая гласит:
| Вероятность быть в состоянии с энергией Е пропорциональна exp(-E/kT). | (4.2) |
Физический смысл этой формулы заключается
в том, что теплота окружающей среды (столкновения с другими молекулами)
в определенной степени (в среднем — пропорционально температуре
среды T) возбуждает нашу молекулу, и это позволяет ей забираться
в область более или менее высоких энергий. Все это мы подробнее обсудим
позже, а пока я позволю себе надеяться, что вы эту формулу помните. Напомню
только, что k — это постоянная Больцмана, а T —
абсолютная температура в градусах Кельвина.
Применительно к интересующему
нас вопросу — как соотносятся вероятности пребывания молекулы
на разных высотах — формула Больцмана сводится к барометрическому
соотношению
| [Вероятность быть на высоте
б] относится к [вероятности быть на высоте
а]
как exp(-Eб/kT) относится к exp(-Eа/kT) , |
(4.3) |
где Eа — энергия
молекулы в состоянии "а" (т.е. "здесь"), Eб
— в состоянии "б" ("на высоте 5 км"), а T — абсолютная
температура (для простоты, будем считать, что с высотой она не меняется).
Из-за силы тяжести энергия молекулы
"здесь" ниже, чем "на высоте 5 км"; так что, согласно Больцману, "на высоте
5 км" молекула будет проводить меньше времени, чем "здесь" [раза в полтора-два
меньше; посчитайте сами при T=300oK,
вспомнив, что E=mgh, где m — средняя масса молекулы
воздуха (»30 дальтон = 30 г/моль), g»10м/с2
— ускорение свободного падения, h»5км
— разность высот, а постоянная Больцмана k равна 1.38x10-23
Джоуля/градус (Дж = кгxм2/сек2
= 0.24 кал) в расчете на частицу, т.е. 2 кал/град в расчете на моль (6x10-23)
частиц. Напомню, что R = 2 (кал/моль)/град. называется "газовой
постоянной"].
То есть молекул на высоте в 5
км будет раза в два поменьше, чем здесь. Точнее — поменьше
в равных единицах объема, например — внутри легких (в
чем легко убедиться, попробовав подышать на разных высотах). Однако в
сумме число молекул в монастыре Далай-Ламы больше, чем в этой комнате:
его монастырь куда больше, чем наша комната. То есть там молекула может
находиться в большем числе положений — для свободно летающей
молекулы воздуха это число пропорционально объему помещения. Физики говорят
в этом случае, что в том монастыре возможно куда больше микросостояний
молекулы, чем в нашей комнате. Поэтому вероятность того, что наша молекула
находится где-то в монастыре Далай-Ламы, относится к вероятности
того, что она находится где-то в этой комнате, как
= [Vбexp(-Eб/kT)] : [Vаexp(-Eа/kT)]
Здесь Vа — объем
суммы состояний "а" ("нашей комнаты"), а Vб
— объем суммы состояний "б" ("его монастыря"). Вспомнив элементарную
институтскую математику, а именно, что V можно
представить как exp(lnV), эту формулу можно переписать как
= [exp(-Eб/kT + lnVб)] : [exp(-Eа/kT + lnVа)] =
= [exp(-(Eб — Txk lnVб)/kT)] : [exp(-(Eа — Txk lnVа)/kT)]
Последняя формула очень напоминает формулу (4.3), формулу Больцмана,
но только она относится не к единице объема, а ко всему объему
системы, и — внимание! — вместо энергий Е
в ней стоят величины E — Txk
lnV. Так вот, величина F = E — Txk
lnV называется свободной энергией нашей молекулы воздуха
при заданном объеме V и температуре T. А величина S
= k lnV называется энтропией нашей молекулы в объеме
V.
Это — в данном конкретном
случае, когда "число доступных состояний" — это просто доступный
для молекулы объем V (Vа
— "нашей комнаты", Vб
— "его монастыря").
В общем случае, энтропия S
просто равна k x
[логарифм числа доступных состояний]. А свободная энергия F
связана с энергией Е, энтропией S и температурой Т
системы согласно общей формуле
| F = E — TS | (4.4) |
Из двух состояний стабильнее, т.е.
вероятнее то, у которого свободная энергия ниже:
| [Вероятность_где-то_в_объеме_"б"]
: [вероятность_где-то_в_объеме_"а"] =
= exp(-Fб/kT) : exp(-Fа/kT) = exp[-(Fб-Fа)/kT] |
(4.5) |
Иными словами, более стабильному состоянию системы отвечает то, где ниже F, а самому стабильному состоянию системы (при заданной температуре и фиксированном объеме этой системы) отвечает минимум свободной энергии F.
Таким образом — "свободная энергия" — это естественное расширение обычного понятия "энергии" на случай, когда система обменивается теплом с окружающей средой. Напомню: если тело не обменивается теплом со средой, его стабильное состояние отвечает минимуму его энергии (или, проще: все, что может упасть — падает). А окружающее тепло делает стабильными определенные флуктуации, прыжки — такие, которые минимизируют свободную энергию тела. То есть приток тепла возбуждает молекулы системы, они начинают захватывать многочисленные состояния с более высокой энергией (т.е. их энтропия растет) — и, в результате, молекулы воздуха летают, а не падают на землю.
Спустимся, однако, с Гималаев к
белкам.
Итак — каков же баланс
энергий, энтропий и свободных энергий в разобранном нами примере образования
водородной связи в белковой цепи? Для полной наглядности, сравним ход процесса
в разных условиях:
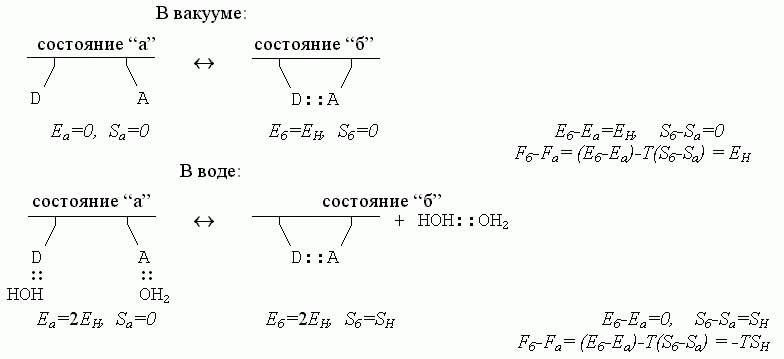
Здесь EH
< 0 - энергия Н-связи, а SH >
0 — энтропия перемещений и вращения свободного тела, т.е. свободной
молекулы Н2О или свободного димера НОH::OH2.
Водородные связи — воды с водой, воды с белком —
стабильны, когда EH -TSH
< 0 (а если EH -TSH
> 0, т.е. если они нестабильны, — то есть в этом случае перед
нами была бы уже не жидкая вода, а пар).
Сравнение приведенных выше схем
показывает, что водородные связи в белковой цепи как бы становятся
— за счет изменения энтропии связывающейся воды — менее
стабильными в водном окружении, чем в вакууме; действительно, в водном
окружении их свободная энергия есть Fб-Fа
= -ТSH , — то есть она
меньше, чем в вакууме, где Fб-Fа=EH.
Еще раз хочу подчеркнуть, что причина этого видимого ослабления — в том, что в водном окружении образовавшаяся внутри белковой цепи Н-связь замещает собой связь цепи с водой. И по той же причине водородные связи, стабилизующие структуру белка в воде носят энтропийную, а не энергетическую природу: энергии двух состояний цепи (с внутрицепочечной связью и без нее) примерно равны, и из этих двух состояний с примерно равной энергией стабильнее та, где выше энтропия, т.е. где больше число микросостояний. А их больше у оторванной молекулы воды, чем у связанной.
Обратите внимание: водородные связи в белковой цепи (в водном окружении) носят энтропийную, а не энергетическую природу именно потому, что энергия Н-связей очень высока: раз так, "свободные" от связей в белке доноры и акцепторы водородных связей в цепи всегда на деле не свободны от всех связей вообще, а связаны с водой. Оторвавшиеся же от белка — при образовании внутрибелковой Н-связи — молекулы Н2О тут же связываются друг с другом, так что энергия компенсируется, и свободно-энергетический выигрыш Н-связей в белке идет только от множественности возможных микросостояний оторвавшихся молекул воды. Правда, чтобы связаться друг с другом, молекулы Н2О должны пожертвовать часть своей обретенной свободы, часть энтропии — но лучше уж потерять небольшую энтропию, чем большую энергию!
То, что (1) молекулы воды сильно
связаны друг с другом водородными связями; и (2) то, что эти связи образуются
только при определенной взаимной ориентации молекул Н2О
— определяет специфику воды как растворителя. Это приводит к разным
интересным эффектам. Подробнее мы об этом поговорим в двух следующих лекциях.